

L’essentiel du PROTOCOLE NATIONAL DE DIAGNOSTIC ET DE SOINS
Document réalisé à partir du PNDS (Protocole National de Diagnostic et de Soins) – Neuropathie amyloïde héréditaire à transthyrétine (NAH-TTR), publié le 19/07/2022 sur le site de Haute Autorité de Santé
ÉDITORIAL
L’amylose héréditaire à transthyrétine (ahTTR) est une maladie génétique rare de l’adulte qui résulte de mutations dominantes sur le gène de la transthyrétine (TTR). On estime qu’il y aurait environ 800 patients symptomatiques en France et environ 3000 porteurs asymptomatiques à risque de développer la maladie.
Il s’agit d’une affection invalidante qui conduit à l’accumulation de protéines TTR malformées dans plusieurs tissus, notamment les nerfs et le coeur. L’atteinte des nerfs conduit à des troubles sensitifs et moteurs des jambes et des bras, l’atteinte du système nerveux autonome à des troubles digestifs, urinaires et sexuels, et l’atteinte cardiaque à un essoufflement et des malaises. La maladie peut aussi toucher les reins, les yeux et le cerveau. Le diagnostic est basé sur la preuve biopsique de l’amylose et sur l’analyse du gène TTR. L’ahTTR est fatale en quelques années en l’absence de traitement.
Au vu de l’atteinte systémique, le suivi est multidisciplinaire et neurologues, cardiologues, gastro-entérologues et rééducateurs prennent en charge conjointement les patients.
Fait rare dans le domaine des maladies génétiques, plusieurs traitements de fond sont aujourd’hui disponibles dans l’ahTTR : certains traitements ciblent la protéine TTR – greffe de foie, tafamidis – et d’autres traitements ciblent l’ARNm TTR hépatique – patisiran, inotersen.
Dans ce contexte, le PNDS rédigé en 2017 méritait d’être révisé et complété avec les données concernant ces nouvelles thérapies issues des biotechnologies. Cette brochure met en exergue les points clés de ce PNDS 2022 révisé.
Pr A. Echaniz-Laguna
Neurologue
Le Kremlin Bicêtre
Mode d’emploi pour la navigation



Définition des acronymes
ARN : acide ribonucléique
ARNi : ARN interférent
ARNm : ARN messager
ASMR : Amélioration du Service Médical Rendu
ASO : oligonucléotide anti-sens
AV : Acuité Visuelle
AVC : Accident Vasculaire Cérébral
ATTR/TTR : (Amylose à) Transthyrétine
BNP : B-type Natriuretic Peptide
CADT: Compound Autonomic Dysfunction Test
ECG : Électrocardiogramme
ENMG : Électroneuromyogramme
ETT : Echographie Trans-Thoracique
FAP : Functional Ambulation Performance
FO : Fond d’Œil
IMC (BMI) : Indice de Masse Corporelle (Body Mass Index)
LAF : Lampe À Fente
MIBG : MetaIodoBenzylGuanidine
NAH : Neuropathie Amyloïde Héréditaire
NIS : Neuropathy Impairment Score.
NYHA : New York Heart Association
PND: Peripheral Neuropathy Disability
SMR : Service Médical Rendu
SNC : Système Nerveux Central
TO : Tonus Oculaire
TH : Transplantation Hépatique
La neuropathie amyloïde héréditaire à transthyrétine (NAH-TTR) est une maladie rare, systémique, de transmission autosomique dominante résultant de mutations sur le gène de la protéine transthyrétine (TTR). Elle ne touche que l’adulte. Les symptômes sont dus à des dépôts extracellulaires de substance amyloïde, principalement dans le système nerveux périphérique (somatique et autonome) et dans le cœur, mais de multiples organes peuvent être atteints, notamment les yeux et les reins.

La prévalence moyenne mondiale de la NAH-TTR est estimée à 1/1 000 000 dans la population générale. Elle peut atteindre 1/1 000 dans les régions endémiques que sont le Portugal, la Suède et le Japon. En France, plus de 500 cas de NAH-TTR sont recensés.
Deux formes cliniques peuvent être distinguées :
- la forme à début précoce : Il s’agit le plus souvent de sujets âgés de 25 à 35 ans d’origine portugaise. Elle concerne 25% des patients en France.
- la forme à début tardif, qui est la majorité des cas en France (75% des patients).La déclaration est tardive (après 50 ans) et les antécédents font défaut dans la majorité des cas.
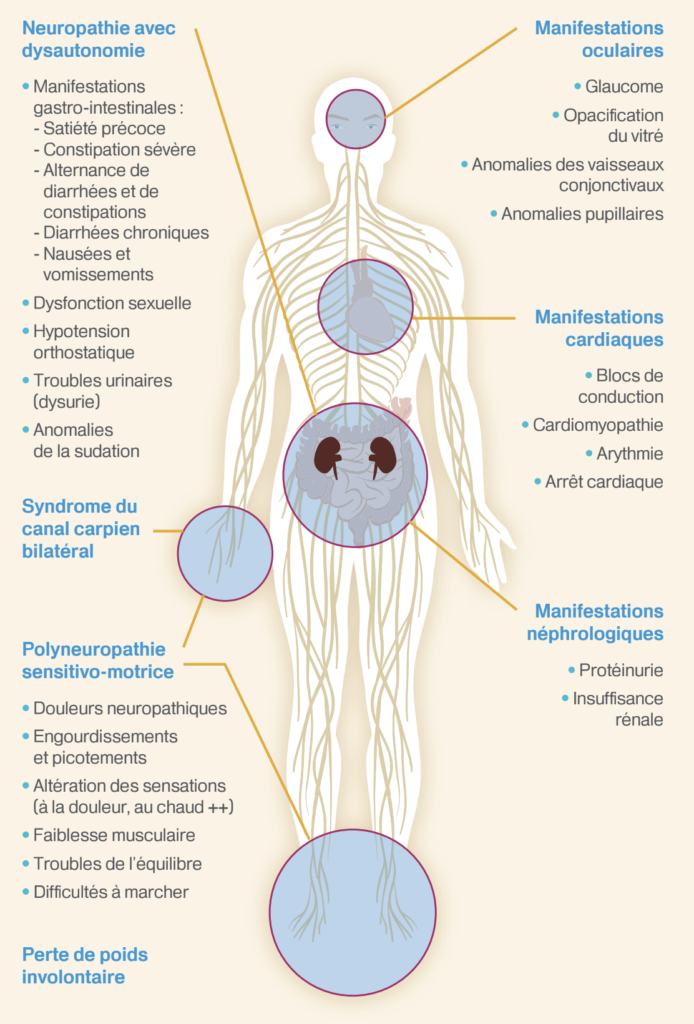

Tableaux qui doivent faire suspecter le diagnostic
- Une polyneuropathie évolutive, sensitive ou sensitivomotrice d’étiologie indéterminée après bilan de première intention
- ou une neuropathie étiquetée neuropathie diabétique, PIDC (polyneuropathie inflammatoire démyélinisante chronique) avec une évolution inattendue par rapport au diagnostic initialement retenu
- a fortiori si :
- présence d’une dysautonomie (troubles érectiles, diarrhée, hypotension orthostatique) sans diabète,
- aggravation rapide du déficit sur quelques mois ou années,
- invalidante avec troubles de la marche en 2-3 ans,
- précédée d’un syndrome du canal carpien bilatéral,
- associée à un amaigrissement important de 10%,
- association à une cardiopathie infiltrative ou à des troubles de conduction sévères.
Y compris chez des sujets jeunes de 30 ans d’origine portugaise avec antécédent familial, mais aussi chez des patients de 50 ans ou plus avec ou sans histoire familiale. La confirmation du diagnostic repose sur :
- la présence d’une mutation amyloïdogène du gène de la TTR,
- la mise en évidence de dépôts amyloïdes sur biopsie tissulaire, immunomarqués pour la transthyrétine.

Test génétique
L’analyse en biologie moléculaire se fait par séquençage du gène de la TTR situé sur le chromosome 18q. Plus de 100 mutations amyloïdogènes de la TTR ont été décrites. La plus fréquente des mutations observées en France est Val30Met rencontrée dans les deux tiers des cas. D’autres mutations plus rares telles que les mutations Ser77Tyr et Ile 107Val peuvent être rencontrées.

En pratique
Après avoir informé le patient et demandé son consentement éclairé, le diagnostic moléculaire est réalisé à partir de l’ADN extrait de son sang ou de sa salive. L’analyse devra obligatoirement être réalisée dans un laboratoire habilité. Le résultat sera ensuite transmis au prescripteur qui rendra son diagnostic au patient.
Si une mutation est révélée, l’enquête familiale doit être systématiquement proposée : elle consiste à dépister les porteurs sains de la famille d’un patient porteur d’une anomalie génétique.
Dans la NAH-TTR, le dépistage est conseillé en raison de la gravité de la maladie, de la disponibilité de traitements efficaces en début de maladie et des implications pour la descendance. Les patients asymptomatiques seront suivis pour identifier au plus tôt le début de la maladie et initier un traitement anti-amyloïde.
Test Anatomopathologique
La mise en évidence histologique de dépôts amyloïdes est essentielle pour le diagnostic d’amylose. Les dépôts sont identifiés par la coloration au Rouge Congo et aspect biréfringent vert pomme sous microscope polarisé par immuno-marquage anti-TTR. L’amylose étant systémique, les dépôts amyloïdes peuvent être mis en évidence sur de nombreux prélèvements, en privilégiant une biopsie non-invasive telle qu’une biopsie des glandes salivaires accessoires.
En pratique
Afin d’optimiser la détection des dépôts amyloïdes, le matériel tissulaire devra être en quantité suffisante (au minimum 3 glandes salivaires par exemple).
Attention : Une biopsie négative n’élimine pas le diagnostic et doit amener à renouveler le prélèvement sur d’autres sites, de préférence « symptomatiques » (tube digestif, rein, cœur).


Il s’agit d’une double annonce : celle de la maladie chronique grave et celle de l’origine génétique. Le médecin se devra de délivrer une information lisible et adaptée à la personne afin de permettre une meilleure adhésion pour la prise en charge. Le médecin expliquera au patient la nécessité de la réalisation de l’enquête génétique familiale et exposera les différents tests qui seront réalisés. Les retombées sur l’existence du patient et de son entourage peuvent être importantes et une écoute active de l’équipe médicale et un accompagnement par un psychologue sont très importants.
Le bilan d’extension comprend des évaluations neurologiques, cardiaques, digestives, ophtalmiques et rénales.
Évaluation neurologique :
L’exploration neurologique comprendra :
- un interrogatoire avec recherche d’une atteinte des petites fibres en utilisant le questionnaire SFN-SIQ (small fiber neuropathy symptoms inventory questionnaire),
- un examen de la force des différentes modalités sensitives et l’analyse des réflexes ostéo-tendineux: items du score NIS (Neuropathy Impairment Score),
- une évaluation de la force de préhension manuelle (Jamar),
- une évaluation locomotrice : scores PND (Tableau 1) & FAP (Tableau 2) et des capacités fonctionnelles : score RODS,
- la dysautonomie sera dépistée avec le score CADT (Compound Autonomic Dysfunction Test) avec recherche d’hypotension orthostatique.

L’électromyogramme
Il viendra compléter l’évaluation clinique à la recherche de la neuropathie sensitivomotrice. Il explorera les 4 membres. D’autres techniques existent pour explorer les petites fibres mais ne sont pas disponibles dans tous les centres : la biopsie cutanée par punch, le sudoscan, les potentiels évoqués laser ou le thermotest.
Évaluation cardiaque
L’atteinte cardiaque est quasi constante au cours d’une NAH-TTR. Elle est souvent « silencieuse », latente et méconnue, alors même qu’elle a une grande importance pour le pronostic.
L’évaluation cardiaque doit permettre :
- d’apporter une information quant au pronostic du patient
- de dépister une dénervation cardiaque sympathique ou parasympathique, très précoce et fréquente
- de détecter une cardiopathie infiltrative (parfois précoce ou isolée)
- de détecter des complications cardiaques qui nécessiteraient une prise en charge spécifique : implantation d’un stimulateur ou d’un défibrillateur cardiaque, dépistage d’une insuffisance cardiaque débutante.
Cette évaluation sera idéalement réalisée dans un centre de référence disposant d’un plateau d’imagerie et d’explorations cardiaques complet. Le diagnostic définitif d’amylose cardiaque reste théoriquement histologique, mais il existe un consensus pour poser le diagnostic positif d’amylose cardiaque quand il existe à la fois une amylose systémique avec preuve génétique et histologique et des signes concordants aux examens cardiaques non invasifs, en particulier l’échocardiographie et la scintigraphie osseuse aux biphosphonates. Ainsi, une fixation cardiaque de ce traceur est quasi-pathognomonique d’une amylose cardiaque.
Pronostic vital
Les facteurs ci-dessous sont associés à un moins bon pronostic :
- Génotypes TTR VAL107 et TTR MET30 avec début clinique tardif.
- Troubles digestifs précoces et IMCm bas.
- Insuffisance cardiaque avec hospitalisation.
- BNP ou NT-pro BNP et troponine augmentés.
- Scintigraphie osseuse aux biphosphonates positive (fixation cardiaque).
- Caractère transmural du rehaussement tardif par le gadolinium à l’IRM cardiaque.
- Présence d’une dysautonomie cardio-vasculaire : hypotension orthostatique et/ou anomalies du test à l’atropine ou de la scintigraphie au MIBG.
Pronostic neurologique
Certains génotypes -TTR V30M tardif & TTR VAL107- sont associés à une atteinte plus précoce de la marche.
Le bilan initial du patient comportera plusieurs explorations et se fera en concertation avec les différents professionnels impliqués. Il servira de référence pour le suivi.
Le bilan de suivi sera dans la mesure du possible standardisé, adapté selon la mutation, le phénotype clinique et le traitement reçu. Il pourra s’enrichir en fonction des plaintes du patient.
LA PRISE EN CHARGE DE LA NEUROPATHIE AMYLOÏDE FAMILIALE1
Les objectifs de la prise en charge thérapeutique sont :
- 1De proposer, dans la mesure du possible, un traitement anti-amyloïde
- 2D'initier, si besoin, des traitements symptomatiques
- 3De prévenir et traiter les défaillances d'organes
- 4De prévenir l’apparition d’autres cas de la maladie dans la famille (conseil génétique).
Traitements anti-amyloïde
Il y a plusieurs traitements anti-amyloïdes visant à ralentir ou à stopper la progression de la maladie en empêchant et/ou réduisant la formation de dépôts d’amylose. Le choix du traitement dépendra du phénotype clinique et du stade de la maladie.
Les traitements sont indiqués uniquement en cas de NAH-TTR symptomatique.

La transplantation hépatique
La transplantation hépatique permet de supprimer la principale source de protéine TTR mutée. Les indications sont exceptionnelles. La transplantation est indiquée uniquement chez le sujet jeune avec mutation Val30Met, après échec des traitements pharmacologiques. La maladie peut continuer à progresser après la TH en raison de la production de TTR « sauvage » amyloïdogène chez les patients à début tardif, la production persistante de TTR mutée dans l’oeil et dans le cerveau et la production de TTR « sauvage » amyloïdogène.


Stabilisateurs du tétramère
Tafamidis 20 mg
Le tafamidis 20 mg est indiqué dans le traitement de la NAH-TTR chez les patients adultes avec une polyneuropathie symptomatique de stade 1 pour retarder le déficit neurologique périphérique.
L’efficacité du tafamidis a été évaluée chez des patients TTR Val30Met à début précoce et à un stade précoce de la maladie. Après 18 mois de traitement par tafamidis, la progression de la neuropathie et la qualité de vie n’étaient pas différents versus placebo. Des analyses complémentaires ne considérant pas le recours à la transplantation hépatique comme un échec ont montré une plus grande proportion de patients répondeurs dans le groupe tafamidis vs placebo. Une étude à long terme sur 5 ans a montré la possibilité de stopper la progression de la maladie en cas de score NIS initial inférieur à 10 mais aussi une progression de la maladie chez les patients avec un score NIS initial de 14. Chez les patients Val30Met à début tardif ou avec une forme avancée, le handicap progresse dans 55% des cas, avec parfois une aggravation de la dysautonomie. Chez les patients non TTR Val30Met, une étude de phase 2 a montré une aggravation clinique à 1 an sous tafamidis.
Au total, les analyses d’efficacité à long-terme du tafamidis ont montré les effets suivants sur la progression de la neuropathie : 1/3 de patients répondeurs, 1/3 de répondeurs partiels et 1/3 de non répondeurs.
Tafamidis 61 mg
Le tafamidis 61 mg est indiqué dans le traitement de l’amylose TTR sauvage ou héréditaire chez les patients adultes avec une cardiomyopathie.
L’efficacité du tafamidis sur la cardiopathie amyloïde (sénile & NAH-TTR) a été évaluée dans l’étude ATTRACT, incluant 441 patients, dont la majorité (76%) avec une forme sénile de la maladie, les autres ayant une forme héréditaire avec variant Val122Ile, Thr60Ala ou Ile68Leu. Une réduction de 13% de la mortalité absolue (soit 31% de réduction de mortalité relative) a été observée sous tafamidis, avec aussi une réduction significative des hospitalisations cardiovasculaires. Le traitement était remarquablement bien toléré.
Dans les analyses de sous-groupe le bénéfice du traitement était particulièrement prononcé chez les patients avec un NYHA Classe II et chez les patients avec une amylose TTR sauvage.
Diflunisal
Le diflunisal est un anti-inflammatoire non stéroïdien. Une étude contre placebo a été réalisée pour évaluer l’efficacité du diflunisal dans la NAH-TTR sur 24 mois chez des patients d’âge divers et avec différentes mutations TTR. Cette étude a montré une réduction de 60% de la progression du score NIS+7 et du score NIS après 2 ans de traitement. Ce médicament n’est pas disponible en France mais il est disponible aux Etats-Unis ainsi que dans certains pays européens : Italie & Suède notamment.
Silençage du gène TTR
Patisiran – ARN interférent
Le patisiran est indiqué dans la NAH-TTR chez les patients avec une polyneuropathie de stade 1 ou 2, avec un service médical rendu (SMR) important et une amélioration du service rendu modérée (ASMR III).
Le patisiran est un petit ARN interférent (ARNi) ciblant l’ARN messager (ARNm) du gène TTR pour inhiber la production de protéine TTR mutée et sauvage au niveau hépatique. Le patisiran a fait l’objet d’une étude de phase III, APOLLO, chez des patients atteints de NAH-TTR stade 1 ou 2 avec des âges de début variables et différentes mutations TTR. La variation du score mNIS+7, était significativement positive après 18 mois de traitement par patisiran avec une amélioration chez 56% des patients versus 4% dans le groupe placebo. L’impact favorable a aussi été démontré sur la qualité de vie (NORFOLK-QoL-DN), la vitesse de marche et le score de dysautonomie COMPASS-31. Le profil de tolérance était acceptable.
Ces données d’efficacité et de tolérance ont été confirmées dans l’étude d’extension à 12 mois.
Inotersen - Oligonucléotique antisens
L’inotersen est indiqué dans la NAH-TTR chez les patients avec une polyneuropathie de stade 1 ou 2, avec un service médical rendu (SMR) important et une amélioration du service rendu mineur (ASMR IV).
L’inotersen est un oligonucléotide antisens (ASO) se liant à l’ARNm TTR pour le dégrader par l’intermédiaire de la RNase H1. L’inotersen a fait l’objet d’une étude de phase III, NEURO-TTR, chez des patients avec NAH-TTR stade 1 ou 2 avec des âges de début variables et différentes mutations TTR. La variation du score mNIS+7 et de l’échelle de qualité de vie NORFOLK-QoL-DN était significative à 15 mois chez les patients sous traitement avec une stabilisation ou amélioration chez 36% des patients sous inotersen versus 19% sous placebo. Le profil de tolérance à court terme était marqué par un risque de thrombopénie potentiellement fatale (1 décès dans l’étude), ainsi que par un risque de glomérulonéphrite.
Les données d’efficacité ont été confirmées dans une étude d’extension, sans augmentation du risque de thrombopénie grave ou de glomérulonéphrite.
| Dénomination Commune Internationale (DCI) | Tafamidis 20 mg2 | Patisiran3 | Inotersen4 |
| Nom commercial* | VYNDAQEL® | ONPATTRO® | TEGSEDI® |
| Mode d’action | Stabilisateur du tétramère de la transthyrétine | ARN interférent Blocage spécifique dans le cytoplasme des hépatocytes de la synthèse de la transthyrétine | Oligonudéotide antisens Blocage non spécifique dans le noyau de la synthèse de la transthyrétine |
| Indication AMM (autorisation de mise sur le marché) | Le tafamidis est indiqué dans le traitement de la NAH-TTR chez les patients adultes présentant une polyneuropathie symptomatique de stade 1 pour retarder le déficit neurologique périphérique. | ONPATTRO est indiqué dans le traitement de l’amylose héréditaire à transthyrétine chez les patients adultes avec une polyneuropathie de stade 1 (marche autonome) ou de stade 2 (marche avec aide). | TEGSEDI est indiqué dans le traitement de la polyneuropathie de stade 1 ou de stade 2 chez les patients adultes atteints d’amylose à transthyrétine héréditaire. |
| Service médical rendu (SMR) | Important | Important | Important |
| Amélioration du service médical rendu (ASMR) | Mineure IV | Modérée III | Mineure IV |
| Intérêt de santé publique (ISP) | Non | Oui | Non |
| Place dans la stratégie thérapeutique | Avis de la Haute Autorité de la Santé (HAS) VYNDAQEL Octobre 2019
VYNDAQEL (tafamidis) reste une option thérapeutique dans la polyneuropathie de stade 1 dans l’hATTR |
Avis HAS ONPATTRO Mars 2019
Patisiran est un traitement de première intention dans le traitement de l’amylose héréditaire à transthyrétine chez les patients adultes avec une polyneuropathie de stade 1 ou de stade 2. |
Avis HAS TEGSEDI Mars 2019
Inotersen est un traitement de deuxième intention, chez les patients qui ne peuvent bénéficier d’ONPATTRO, dans le traitement de l’amylose héréditaire à transthyrétine, chez les patients adultes avec une polyneuropathie de stade 1 ou de stade 2. |
| Dénomination Commune Internationale (DCI) | Tafamidis 61 mg2 |
| Nom commercial* | VYNDAQEL® |
| Mode d’action | Stabilisateur du tétramère de la transthyrétine |
| Indication AMM (autorisation de mise sur le marché) | VYNDAQEL est indiqué dans le traitement de l’amylose à transthyrétine de type sauvage ou héréditaire chez les patients adultes avec une cardiomyopathie. |
| Service médical rendu (SMR) | Important |
| Amélioration du service médical rendu (ASMR) | Important II |
| Intérêt de santé publique (ISP) | Oui |
| Place dans la stratégie thérapeutique | Avis HAS VYNDAQEL 61 mg Octobre 2020
VYNDAQEL (tafamidis) est un traitement de 1ère intention de l’amylose à transthyrétine avec cardiomyopathie. |
De nombreux symptômes peuvent altérer la qualité de vie des patients :
- Des manifestations neurologiques, avec des douleurs neuropathiques, des troubles végétatifs qui peuvent être digestifs (hauts ou bas), urinaires, sexuels ou bien encore une hypotension orthostatique.
- Des complications en lien avec l’atteinte d’un organe : cardiaques, ophtalmologiques et rénales. Il conviendra d’apporter à chaque patient un traitement spécifique à ses manifestations.
- Kinésithérapie : bénéfice démontré de l’activité physique sur la masse musculaire, la force musculaire et la capacité de marche dans la NAH-TTR.
- Ergothérapie : envisagée dès qu’il existe un retentissement sur l’autonomie : toilette, habillage, alimentation, écriture, utilisation d’un clavier d’ordinateur.
- Diététique et nutrition : en cas de dénutrition, confirmée par la diminution de l’albumine et la préalbumine*, des apports hyperprotéiques peuvent être prescrits.
- Psychothérapie : un soutien psychologique peut être proposé sous forme d’entretiens individuels, de groupe de paroles ou d’ateliers d’éducation thérapeutique.
- Prise en charge médico-sociale.
- Education thérapeutique : des programmes d’éducation thérapeutique, ont été développés par le CHU de Bicêtre (édAmyl) et l’Association Française contre l’amylose (ORIGAMY) pour répondre au besoin des patients atteints d’amylose.
- Les professionnels de santé, les patients et les aidants doivent être informés de l’existence d’une association de patients : association française contre l’amylose (https://amylose.asso.fr/).
* Attention à l’interprétation de la préalbumine dont le résultat est faussé chez les patients sous silençage.
La prise en charge globale du patient repose sur une coopération pluridisciplinaire qui peut être coordonnée par un médecin du centre national de référence ou d’un centre de la filière FILNEMUS, en lien avec les autres spécialistes (neurologues, cardiologues, ophtalmologistes...) et les équipes paramédicales. Un programme d’éducation thérapeutique peut également être proposé au malade. De plus, il est également important de l’informer de l’existence d’une association de patients : l’association française contre l’amylose.
CENTRES DE RÉFÉRENCES
DES NEUROPATHIES AMYLOÏDES FAMILIALES ET AUTRES NEUROPATHIES RARES (NNERF)
Le centre de référence national des neuropathies amyloïdes familiales et autres neuropathies périphériques (CHU Bicêtre) et les centres d’expertises dans la prise en charge de l’amylose hATTR peuvent vous aider dans votre démarche de diagnostic et suivi.

Site coordonnateur
APHP- Kremlin Bicêtre
Pr Andoni Echaniz-Laguna
www.nnerf.org
Pour connaître la liste des centres habilités à réaliser le test de dépistage, rendez-vous sur le site orpha.net
Le PNDS (Protocole National de Diagnostic et de Soins) – Neuropathie amyloïde héréditaire à transthyrétine (NAH-TTR) a été coordonné par le Pr Andoni Echaniz-Laguna, Coordonnateur du CRMR National « Neuropathies Périphériques Rares » du CHU de Bicêtre.
1. PNDS NAH-TTR. Centre de Référence CHU Bicêtre. Juillet 2022.
2. HAS. Avis CT de VYNDAQEL 20 mg, 2019
3. HAS. Avis CT de ONPATTRO, 2019
4. HAS. Avis CT de TEGSEDI, 2019
5. HAS. Avis CT de VYNDAQEL 61 mg, 2020